Our Research
Artificial Intelligence in Next-generation Biology and Medicine
Research overview of the Wang Genomics Lab
We develop novel genomics and bioinformatics methods to improve our understanding of the genetic basis of human diseases.
The research in our laboratory aims to develop novel genomics and bioinformatics methods to improve the diagnosis, treatment, and prognosis of rare diseases, to ultimately facilitate the implementation genomic medicine on scale. Our research can be divided into several areas.
AI in Medicine: we are developing machine-learning and artificial intelligence (AI) approaches from clinical phenotypic information in Electronic Health Records (EHR) to correlate genotype and phenotype together, and better understand the phenotypic heterogeneity of inherited diseases. We are especially interested in transformer models, including BERT, GPT and other similar models, for the analysis of multimodal data. Some examples of computational tools that we developed include Phenolyzer, EHR-Phenolyzer, Phen2Gene, PhenoGPT and GestaltMML, which use natural language processing and multimodal machine learning to predict possible genetic syndromes and candidate genes.
Long-read Sequencing: we are also developing genomic assays and methods to analyze long- read data, such as those generated from PacBio and Oxford Nanopore long-read sequencing. These methods help identify causal genetic variants on cases that failed to be diagnosed by traditional whole genome/exome sequencing approaches, and help map aberrant DNA modifications such as methylations in tissues from patients in comparison to controls. Some examples of computational tools developed by our lab include RepeatHMM, LinkedSV, NanoMod, NanoRepeat, ContextSV, LongReadSum, NanoCaller, LIQA, DeepMod and DeepMod2 and SCOTCH.
Genome Reinterpretation: we are developing analytical pipelines for whole genome and whole exome sequencing data, all the way from FASTQ/FAST5/POD5 files to biological insights. Some examples of computational tools used in the lab include ANNOVAR, Phenolyzer, InterVar, CancerVar and PhenoSV. These approaches facilitate a better understanding of the functional content and clinical insights from sequencing data, and use genome reinterpretation to facilitate precision genomic medicine.
AI in Medicine
We develop and apply deep learning based natural language processing approaches to mine a gold mine of clinically relevant information from electronic health records.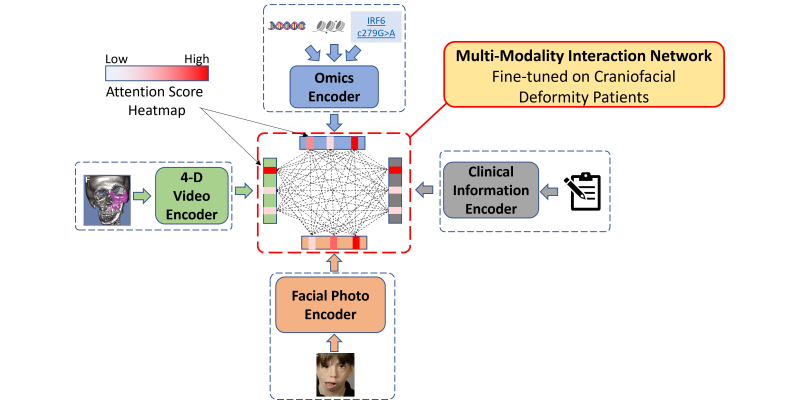
Unlike the massive amounts of texts from Twitter, Reddit and Facebook with questionable quality, clinical notes in electronic health records (EHRs) in our hospital system represent expert-compiled and human curated information on different diseases. These billions of records are sitting there, waiting to be explored by the next generation of data scientists. By demonstrating clinical utility of EHR-Phenolyzer with Dr. Chunhua Weng, we are among the pioneers to integrate deep phenotyping and clinical sequencing data for patients with rare diseases, to facilitate diagnosis and to improve decision making in patient care. Recent development of deep learning based NLP in the past few years (such as BERT/GPT) revolutionalized the entire field, such that machines can finallly understand human logic and human thinking when writing clinical notes for patients. We leveaged these latest development in the field, and use clinical phenotypic data to inform the prioritization of variants, and for systematically build knowledgebases on every human disease.
To enable AI in medicine, we are developing and validating scalable approaches to abstracting characteristic phenotypes of all genetic disorders from EHR narratives and standardize the concept representations of these EHR phenotypes. We developed Doc2HPO, that automatically converts a clinical note into computer-understanble terminologies, as well as Phen2Gene, that automatically predict likely genetic syndromes from these phenotypic terminologies. We also build PhenCards as a one- stop shop that catalog all known biomedical knowledge related to clinical phenotypes. With advanced NLP methods, we are also building Knowledge Graphs for all emerging diseases such as COVID-19, so that researchers can quickly gain biological insights from thousands of manuscripts within a few hours by asking computers to read all these papers. We also developed PhenoGPT and PhenoGPT2 for automated extraction of clinical phenotypes from clinical texts. Based on our recent development of GestaltMML, we will develop an ethically focused and data-driven multimodal machine learning algorithm that integrates frontal facial photos, clinical notes, patient demographics, and structured phenotypes to improve diagnosis of genetic syndromes. To incorporate multiomics data into clinical practice, we will develop a multimodal algorithm that takes 4D videos, facial photos, electronic health records, and omics information (genome, transcriptome and epigenome), to prioritize genes in key pathways that are relevant to phenotypic features of craniofacial abnormalities.
long-read sequencing
We develop novel methods and software tools for long-read sequencing data on diverse technical platforms, such as PacBio and Oxford Nanopore.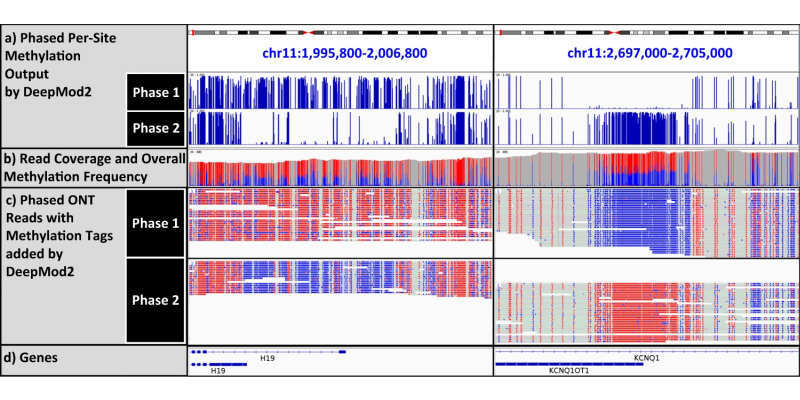
We and others have shown that long-read sequencing technologies, such as Oxford Nanopore and Pacific BioSciences, have revolutionalized the field of biomedical research and genomic medicine, with significant advantages compared to conventional short-read sequencing technologies. For example, long- read sequencing can identify pathogenic SVs missed by short-read sequencing, and even detect those traditional “unsequenceable” SVs. Furthermore, long-read sequencing can detect DNA and RNA modifications directly, contributing to our understanding of the epigenetic regulation of human genome and transcriptome. We are developing various deep neural network approaches to handle unique challenges in long-read sequencing data, to facilitate novel genetic discoveries and accelerate the implementation of precision medicine.
Some examples of the tools that we developed include RepeatHMM, NextSV, LinkedSV, NanoMod, DeepMod, DeepRepeat and NanoCaller. For example, DeepMod is a bidirectional recurrent neural network (RNN) with long short-term memory (LSTM) to detect DNA modifications using raw electric signals of Oxford Nanopore sequencing. DeepRepeat convert a DNA repeat unit and its upstream and downstream units into RGB (red- green-blue) channels of a color image represented by different pixels, thus we transform the repeat detection problem into an image recognition problem by deep neural networks. NanoCaller detects SNPs/indels by examining pileup information from other candidate SNPs that share the same long reads solely using long- range haplotype information in a deep neural network. What is exciting is that we recently won the PrecisionFDA Variant Calling Challenge on MHC (the most difficult region in the entire human genome) using NanoCaller-based Ensemble Caller.
Finally, we have the sequencing platform at the Wang Genomics lab and we are evaluating the use of long-read sequencing to study microbial communities, and demonstrated how this technology can significantly improve our understanding of microbiome and human health. We are applying the method to transcriptome (RNA-Seq) data and demonstrated superior performance in finding differential isoform usage and finding novel fusion genes in various types of cancer. We completed a study on sequencing HTT gene in 1000 patients with Huntinogton's disease and demonstrated the utility of long-read sequencing and long-range haplotype phasing to design allele-specific genome editing strategies. In 2024, we are among the first groups to demonstrate the technical feasibility to generate and analyze single-cell long-read RNA-Seq data using 10X Genomics and Nanopore sequencing, and using Parse Bioscience and Nanopore sequencing. We are super excited and fully prepared to embrace this new frontier and the revolution of long-read sequencing!
Genome Reinterpretation
We develop fully automated pipelines for genome and exome sequencing analysis: all the way from raw sequencing data to biological and clinical insights.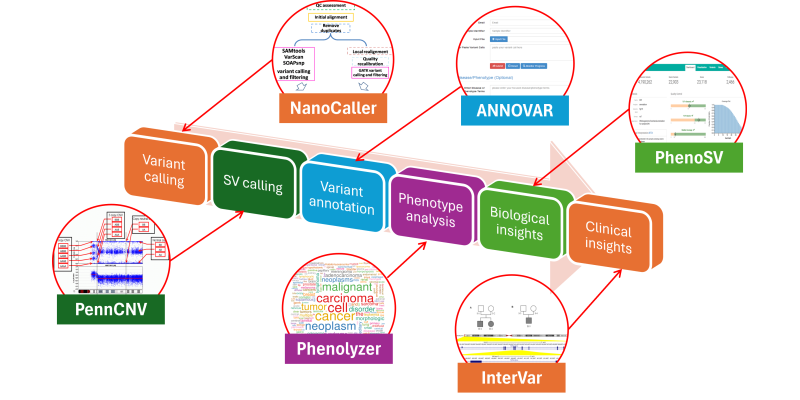
A new generation of sequencing platforms that perform high- throughput sequencing have now made it possible to generate enormous amounts of DNA sequence data on individual human genomes. We develop a suite of bioinformatics methods to understand the functional contents and clinical insights from personal genome sequencing data. Our tools are widely used by research labs, clinical diagnostic labs and pharmaceutical companies to understand genome sequencing data.
Some examples of the computational tools that we created include: PennCNV, ANNOVAR, Phenolyzer, InterVar/wInterVar, EHR-Phenolyzer and CancerVar. For example, our flagship tool, ANNOVAR, is an efficient software to utilize up-to- date information to functionally annotate genetic variants detected from diverse genomes (including human, mouse, worm, fly and many others), and has been widely used by the community, with over 10000 citations. Its web-service version, wANNOVAR, has processed over 300,000 genome/exome files from submissions worldwide as of early 2021. Its commercial version was licensed by Qiagen with a video tutorial. We also developed PhenoSV, a phenotype-aware machine-learning model that interprets all major types of SVs and genes affected. PhenoSV segments and annotates SVs with diverse genomic features and employs a transformer-based architecture to predict their impacts under a multiple-instance learning framework. The combination of these tools enables end-to-end, fast and reliable whole-genome/exome sequencing data analysis on personal genomes, and facilitaets the implementation of genomic medicine on scale.